Werner Syndrome
Background
Otto Werner originally defined Werner syndrome (WS) in 1904 on the basis of sclerodermalike, thin, tight skin and bilateral cataracts. WS is also known as progeria adultorum, progeria of the adult, and pangeria. WS is the most common of the premature aging disorders. Progeria can also refer to Hutchinson-Gilford syndrome, which is described as a lamin A gene defect and has onset early in life. The term progeria is derived from a Greek term meaning "before old age." See the images below.
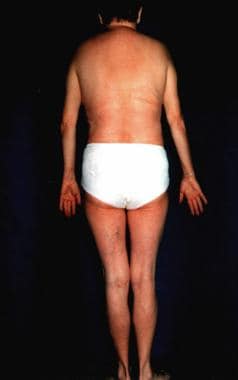 Werner syndrome.
Werner syndrome.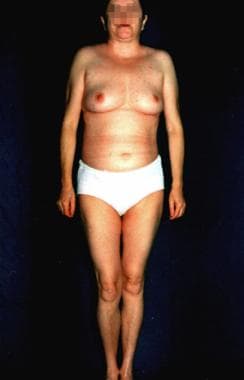 Werner syndrome.
Werner syndrome.The aging process involves increasing errors in the mitotic machinery of dividing cells in the postreproductive stage of life; therefore, WS serves as a model for studying the aging process in vivo and in vitro.[1] However, many organs in patients with WS prematurely undergo changes that are usually associated with aging. No mental retardation is observed.
WS is a premature aging disorder that may serve as a model of normal human aging.[2] In vivo aging is linked with accelerated aging of fibroblasts in culture, possibly due to the genomic instability, a hallmark of WS. Genome instability activates stress kinases, implying that kinase inhibitors may form the basis of antiaging therapies for individuals with WS.
Pathophysiology
Werner syndrome (WS) is an autosomal recessive disorder that affects connective tissue throughout the body. This segmental progeroid syndrome is caused by null mutations at the WRN locus,[3] which codes for a member of the RecQ family of DNA helicases. Enzymes in this group unwind double helix RNA and DNA. The protein is likely to be involved in the response to DNA damage during replication, as well as in the replication and transcription processes. The disease is connected with excessive synthesis of collagen types I and III, which is dependent on elevated messenger RNA (mRNA) levels.
More than 70 disease-causing mutations have been described, the majority being stop codon mutations, splice mutations, or small ins/del-producing truncations of the protein and/or non-sense–mediated decay of mutant mRNA.[4] Two novel WRNmutations were described in patients of South Asian ancestry.[5] Another patient had a homozygous novel 1bp-deletion in exon 19 of WRN, 2426/27delG, causing frameshift and protein truncation R809SfsX2.[6]
The collagenase level is also increased several times. However, a child may have characteristics of WS but with no identifiable mutation in the WRN gene. Such cases are classified as atypical WS.[7] A paper from 2010 reported 18 new mutations, including 2 genomic rearrangements, a deep intronic mutation resulting in a novel exon, a splice consensus mutation leading to utilization of the nearby splice site, and 2 rare missense mutations.[8]
WS have been classified is a member of the RecQ family of DNA helicases implicated in the resolution of DNA structures leading to the stall of replication forks.[9] Fragile sites may be DNA regions particularly sensitive to replicative stress. Evidence was recently presented of a crucial role for a helicase in protecting cells against chromosome breakage at normally occurring replication fork-stalling sites.
Human progeroid syndromes are linked with mutations in single genes accelerating some, but not all, features of normal aging.[10] Most are associated with defects in genome maintenance.
LMNA (lamin A/C) gene mutation has been described with atypical Werner syndrome, with the severe metabolic complications, the extent of the lipodystrophy being linked with A133L mutation in the LMNA gene.[11, 12]
Epidemiology
Frequency
United States
WS is a rare disorder. The estimated incidence is 1 case in 1 million individuals.
International
WS is more common in Japan and Sardinia than in other regions.[13] About 1000 cases are reported in the world; more than 800 of these cases are in Japan.
Race
No racial predilection is reported.
Sex
Both sexes are affected equally.
Age
The onset of WS might occur in individuals in their mid teens, or it may be delayed until an individual is as old as 30 years. The average age of patients at the time of diagnosis is 37 years.
History
In young adults, mutation in the WS gene is believed to be associated with clinical symptoms typically found in elderly individuals.
The most important feature of WS is healthy development in the patient's first decade of life.
Adult progeria is usually diagnosed on the basis of characteristic clinical features and typical concomitant diseases.
The hallmark of this syndrome is a striking disproportion between the patient's real age and the patient's appearance.
In general, this is an adult-onset disorder, with the earliest sign the lack of a growth spurt during adolescence.[4] A prematurely aged appearance with gray hair and sclerodermatous cutaneous changes begins in the 20-30s in association with cataracts, diabetes mellitus, atherosclerosis, cancers, and osteoporosis.
Metabolic abnormalities, including insulin resistance, may not be a pivotal part at disease onset.[14]
Physical
Perform a thorough clinical and laboratory examination, keeping in mind the patient's increased risk of neoplasms.
Characteristic clinical features of the disease
General appearance is as follows:
- Short stature, usually less than 1.60 m, is observed.
- Thin skin is present on the acral surfaces.
- Muscle atrophy is noted.
- The skull is relatively large, with a disproportionate lower part of the face.
Skin findings are as follows:
- Wrinkling and aging of the face occurs.
- A sclerodermalike appearance with nose and lip atrophy is typical.
- The nose is pinched, and the cheeks are sunken because of fat loss, which causes the birdlike facial appearance.
- Loss of subcutaneous fat complicated with ulceration can be observed on the shins and feet.
- In most patients, calluses, hyperkeratosis, and ulcerations on the soles are present mainly over bony prominences.
Graying of the hair, loss of hair, and nail dystrophy usually are observed.
A high-pitched voice is characteristic.
Related diseases
Cataracts, as follows:
- Rapidly progressing cataracts typically occur when patients are aged 20-40 years.
- Cataracts are usually posterior and subcapsular.
- Osteoporosis: Disturbances in the parathyroid glands are the main cause of osteoporosis.
- Diabetes mellitus
Neoplasia, as follows:
- Hematologic malignancies (leukemia and preleukemic conditions of the bone marrow [15] )
- Carcinomas of the thyroid [15] and other organs
- Sarcomas (soft tissue [15] )
- Meningiomas [15]
- Cutaneous malignancies, including malignant melanoma [15]
- Possible evolution of ankle and heel ulcers into squamous cell carcinomas
- Hyperthyreosis
- Pituitary dysfunction
- Vascular changes (arteriosclerotic type)
- Hypogonadism or agonadism and premature menopause
- Soft tissue calcification
- Primary bone neoplasms [15]
Causes
WS is a genetic disorder. See Pathophysiology.
Differential Diagnoses
Laboratory Studies
Perform a thorough clinical and laboratory examination, keeping in mind Werner syndrome (WS) patients' increased risk of neoplasms.
WS has no specific laboratory abnormalities. Any laboratory abnormalities are related to concomitant diseases, especially diabetes mellitus, arteriosclerosis, and hypogonadism, which are often seen in WS.
One should perform the following:
- Fasting blood glucose test
- Oral glucose tolerance test
- Triiodothyronine, levothyroxine, and thyrotropin tests
- Appropriate vascular studies
Diabetes mellitus is of the late-onset type.
One should check the blood glucose level.
An oral glucose tolerance test should be performed.
Atherosclerosis should be evaluated with a lipid profile.
An elevated low-density-lipoprotein cholesterol level and a high triglyceride level associated with a low high-density-lipoprotein cholesterol level are the risk factors for atherosclerotic vascular disease.
The National Cholesterol Education Program has issued guidelines for the diagnosis and optimal treatment of hyperlipidemia.
Hypogonadism is usually due to intestinal fibrosis.
One should check estradiol, progesterone, and luteinizing hormone/follicle-stimulating hormone (LH/FSH) levels in the sera of female patients.
One should check testosterone and LH/FSH in the sera of male patients.
Other Tests
Both genetic testing and clinical criteria may not establish the diagnosis is some people. Gene testing can be salient to confirm autosomal recessive inheritance.[4]
Histologic Findings
The skin and subcutaneous tissue of the extremities, especially where it is taut on clinical examination, shows epidermal thinning, loss of rete ridges, and dermal fibrosis with or without collagen hyalinization. Pilosebaceous structures are not well formed. Newly synthesized hyalinized collagen tends to replace the subcutaneous fat. No inflammatory infiltrate is usually evident.
Medical Care
Specific treatment for Werner syndrome (WS) does not exist. Only symptomatic treatment of related disturbances is recommended. Results of a 2010 study on mice suggest that vitamin C supplementation could be beneficial for patients with WS.[16]
Consultations
Consultations are related to the concomitant diseases.
Medication Summary
No specific treatment exists.
Complications
Complications of Werner syndrome (WS) are related to the concomitant diseases and symptoms described above (see Physical). Pregnancy complicated by the presence of Werner syndrome has been reported.[17]
The variant genotype of WRN Leu1074Phe has been associated with a 1.36-fold significantly increased risk of breast cancer in Chinese women, particularly pronounced in those carrying Phe/Phe genotype and having an earlier age at menarche, to produce a 3.58-fold increased risk of breast cancer.[18]
Patients with Werner syndrome may develop severe cystoid macular edema after laser therapy.[19]
Prognosis
The prognosis is unfavorable. The mean survival for patients with WS is 46 years. Death usually occurs when patients are aged 30-50 years because of atherosclerosis or malignant tumors. Adroit medical management may enhance life expectancy; one patient was described who survived until dying of acute heart failure at age 68 years.[20]
나이에 비해 일찍 늙는 ‘베르너 증후군’에 걸린 장미향(43) 씨의 사연이 방송을 통해 소개되면서 안타까움을 더하게 했다.
6일 SBS `순간포착 세상에 이런 일이`에서는 2주 전에 이어서 3번째 이야기로 장 씨의 정밀검사 과정이 공개됐다.

‘24kg의 아내’로 불리는 장 씨는 8번 염색체 이상으로 일찍 늙는 병인 베르너 증후군으로 의심됐다. 하지만 여러 병원에서 제대로 된 병명조차 확인할 수 없는 상태였다.
이에 제작진의 도움으로 서울시의 한 종합병원에 옮겨 1주일간 전문의들의 검진을 받게 됐다.
검사결과 의료진은 “근본적인 유전자 검사 결과가 나와야겠지만 ‘베르너 증후군’으로 의심된다”고 밝혔다. 당뇨병으로 나타나는 질병도 이로 인해 파생된 현상이라고 말했다.
한편 아내의 병명을 전해들은 남편 박상기(52) 씨는 “뭐라고 말해야 할지 모르겠다”면서도 “걱정마라. 내가 하라는 대로 하면 좋아질 수 있다"며 아내를 격려했다.
장 씨가 남편을 위해 직접 쓴 쪽지도 시청자들의 눈시울을 붉히게 만들었다. 장 씨는 “눈이 잘 보이고 몸이 나으면 남편이 못하는 일을 도와줄 수 있다고 맹세한다. 죽도록 남편을 사랑한다”고 말했다.
두 부부의 안타까운 사연을 접한 네티즌은 “희망을 잃지 말고 살아가면 좋겠다” “두 사람이 사랑하는 광경이 너무 감동적이다. 빨리 회복되기를 바란다”며 쾌유를 기원했다.




